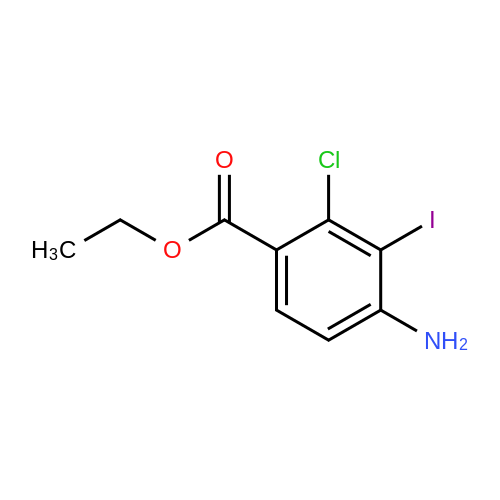
[ 256935-85-0 ] Synthesis Path-Downstream 1~50
- 1
-
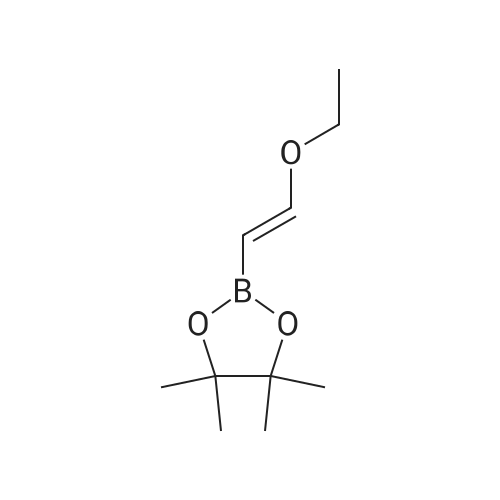
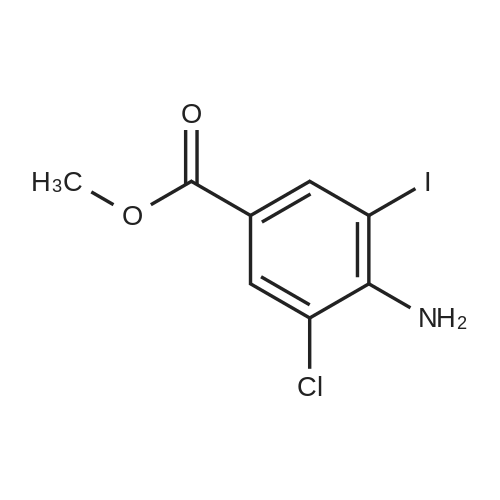
 [ 935676-16-7 ]
[ 935676-16-7 ]

-
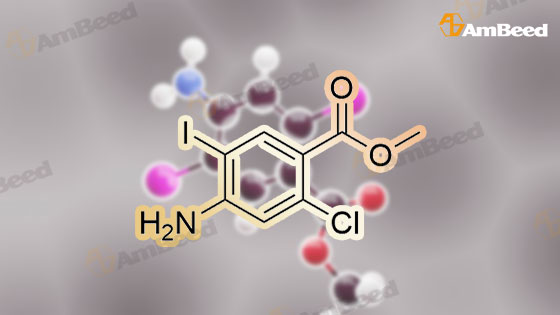
 [ 256935-85-0 ]
[ 256935-85-0 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With hydrogenchloride In ethanol |
|
Reference:
[1]Chen, Yihui; Shibata, Masayuki; Rajeswaran, Manju; Srikrishnan, Thamarapu; Dugar, Sundeep; Pandey, Ravindra K.
[Tetrahedron Letters, 2007, vol. 48, # 13, p. 2353 - 2356]
- 2
-
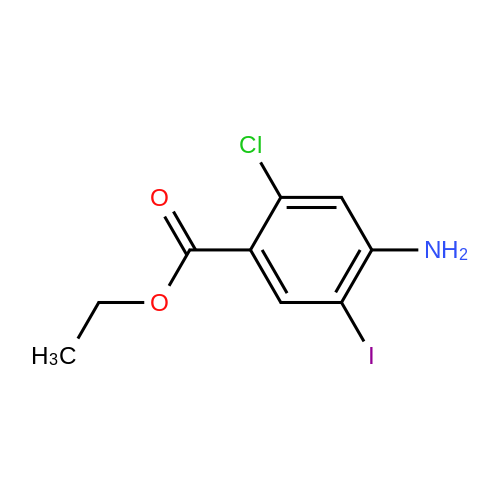
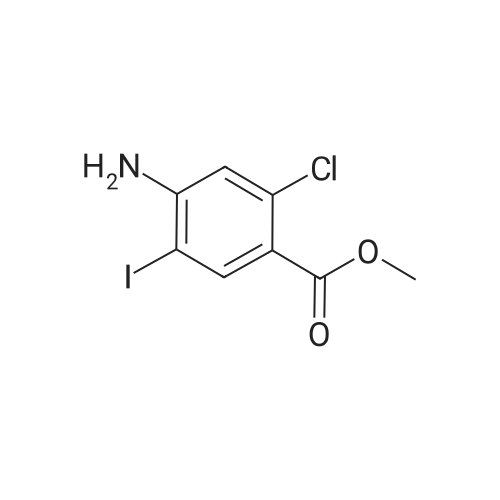
 [ 46004-37-9 ]
[ 46004-37-9 ]
-
methyl 4-amino-2-chloro-3-iodo-benzoate
[ No CAS ]

-
[ 256935-85-0 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With Iodine monochloride; calcium carbonate; In methanol; dichloromethane; at 20℃; for 1.5h; |
B. Methyl 4-amino-2-chloro-5-iodo-benzoate, 23c. To a suspension of compound 23b (1.18 g, 6.38 mmol) and CaCO3 (12.8 mmol, 1.28 g) in MeOH (13 mL) was added a solution of iodine monochloride (6.70 mmol, 1.09 g) in CH2Cl2 (6 mL) dropwise at room temperature. The resulting reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was concentrated and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 20-25% EtOAc/hexanes) to provide methyl 4-amino-2-chloro-5-iodo-benzoate 23c as the major the product and methyl 4-amino-2-chloro-3-iodo-benzoate 23d as the minor product. |
|
With Iodine monochloride; calcium carbonate; In methanol; dichloromethane; at 20℃; for 1.5h; |
To a suspension of compound 23b (1.18 g, 6.38 mmol) and CaCO3 (12.8 mmol, 1.28 g) in MeOH (13 mL) was added a solution of iodine monchloride (6.70 mmol, 1.09 g) in CH2Cl2 (6 mL) dropwise at room temperature. The resulting reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was concentrated and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 20-25% EtOAc/hexanes) to provide methyl 4-amino-2-chloro-5-iodo-benzoate 23c as the major the product and methyl 4-amino-2-chloro-3-iodo-benzoate 23d as the minor product. |
|
With Iodine monochloride; calcium carbonate; In methanol; dichloromethane; at 20℃; for 1.5h; |
To a suspension of compound 22j (1.18 g, 6.38 mmol) and CaCO3 (1.28 g, 12.8 mmol) in MeOH (13 mL) was added a solution of iodine monochloride (1.09 g, 6.70 mmol) in CH2Cl2 (6 mL) dropwise at room temperature. The resulting reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was concentrated and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 20-25% EtOAc/hexanes) to provide methyl 4-amino-2-chloro-5-iodo-benzoate 22k as the major product and methyl 4-amino-2-chloro-3-iodo-benzoate 221 as the minor product. |
|
With iodine; silver sulfate; In ethanol; for 0.25h;Inert atmosphere; |
To a suspension of <strong>[46004-37-9]methyl 4-amino-2-chlorobenzoate</strong> (55.8 mmol) in EtOH (558 mL) was added silver sulfate (55.8 mmol) and iodine (58.6 mmol) under argon. The mixture was stirred for 15 min, filtered and the filtrate was concentrated in vacuo. The residue was partitioned between DCM and a 1 M aq. solution of NaOH. The organic phase was washed with a 1 M aq. solution of NaOH, dried over MgS04 and concentrated in vacuo. The crude was purified by CC (SNAP KP-SIL from Biotage) using Hept/EtOAc/MeOH from 89/11/1 to 81/19/1 to give the mixture of regioisomers as salmon solid. The mixture was enriched from 59 to 66% in methyl 4-amino-2-chloro-3-iodobenzoate by recrystallisation in Hept/EtOAc 75/25, separation of the solid methyl 4-amino-2-chloro-5-iodobenzoate by filtration and evaporation of the mother liquid. LC-MS (B): tR = 0.72 min; [M+CH3CN+H]+: 352.79 In addition, pure methyl 4-amino-2-chloro-5-iodobenzoate regioisomer was isolated as pink to orange solid. LC-MS (B): tR = 0.75 min; [M+CH3CN+H]+: 352.80 |

Reference:
[1]Tetrahedron Letters,2007,vol. 48,p. 2353 - 2356
[2]Patent: US2012/58986,2012,A1 .Location in patent: Page/Page column 33
[3]Patent: US2012/77797,2012,A1 .Location in patent: Page/Page column 33; 34
[4]Patent: US2012/101081,2012,A1 .Location in patent: Page/Page column 53
[5]Patent: WO2014/97140,2014,A1 .Location in patent: Page/Page column 68
- 3
-
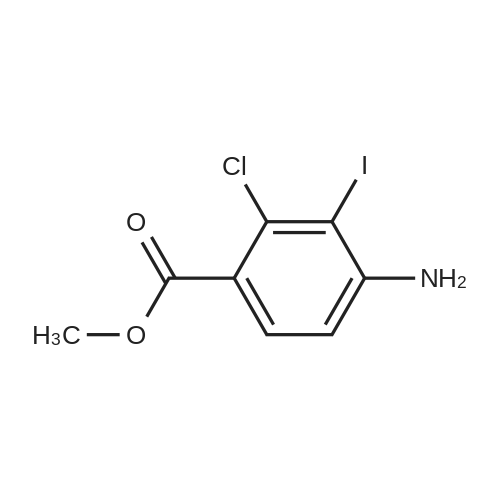
[ 46004-37-9 ]
-
[ 256935-85-0 ]
| Yield | Reaction Conditions | Operation in experiment |
| 56% |
With sodium periodate; iodine; In DMF (N,N-dimethyl-formamide); at 50℃; for 2h; |
To a solution of <strong>[46004-37-9]methyl 2-chloro-4-aminobenzoate</strong> (109 g, 587 mmol) in 440 mL DMF was added NaIO4 (50 g) and iodine (119 g). The mixture was warmed at 50 C. for 2 h. It was then cooled and diluted with water and filtered. The solid was treated with sodium hydrogensulfite (19 g) in water (110 mL) at 40 C. for 1 h. The mix was filtered again and washed with water, followed by isopropanol. Upon drying, the product was obtained in 56% yield. M+H+(312). |
| 17 g |
With Iodine monochloride; calcium carbonate; In methanol; dichloromethane; at 20℃; for 5h; |
To a suspension of methyl 4-amino-2- chlorobenzoate (20.0 g, 107.75 mmol) and CaCO3 (21.58 g, 215.5 mmol) in MeOH (216 mL) was added a solution of iodine monochloride (20.0 g, 123.2 mmol) in CH2CI2 (102 mL). The resulting reaction mixture was stirred at room temperature for 5 h, and quenched by adding cooled water (700 mL) and ethyl acetate (700 mL). It was filtered through celite, the filtrate was treated with 300 mL of 10% sodium thiosulfate, partitioned, and the aqueous layer was extracted with ethyl acetate (400 mL). The combined organic layers were washed with 10% sodium thiosulfate (2x), and brine. The organic layer was dried over Mg504, and concentrated to dryness under vacuum. The residue was adsorbed in silica gel, loaded into flash column which was eluted with 5, 7.5, and 10% ethyl acetate/toluene to afford 17.0 g of title compound as a yellow solid. 1H NMR (400 MHz, CDCI3): 6 8.26 (s, 1H),6.75 (s, 1H), 4.52 (bs, 1H), 3.87 (s, 3H). LCMS (M+1) 312.0, 314.0. |
| 5.86 g |
With sodium periodate; iodine; In N,N-dimethyl-formamide; at 65℃; for 2h; |
Step 1: A stirred solution of compound 6 (5.00 g, 26.9 mmol) in N,N- dimethylformamide (DMF: 18.9 mL) was treated with sodium periodate (2.30 g, 10.8 mmol) and iodine (5.50 g, 21.6 mmol) at room temperature. The resulting brown solution was heated to 65C for 2 hours, and then allowed to cool to room temperature. The reaction mixture was then poured slowly into water (150 mL) with rapid stirring, and the resulting dark brown precipitate was collected by filtration (washing with water) and dried under suction. The resultant dark brown solid was added to 5% aqueous sodium metabisulfite solution with stirring, and the resulting suspension heated at 45-50C for 30 minutes. The brown solid was then collected by filtration (washing with water) and dried under suction. The solid was triturated with isopropanol (30 mL), with stirring, for 30 minutes, and was then filtered and dried under suction to afford compound 7 (5.86 g) as a brown solid. 1H NMR (400 MHz, CDC13) delta 8.26 (s, 1H), 6.75 (s, 1H), 3.87 (s, 3H) |
Reference:
[1]Organic Preparations and Procedures International,2007,vol. 39,p. 90 - 93
[2]Patent: US2004/142940,2004,A1 .Location in patent: Page/Page column 13-14
[3]Tetrahedron Letters,2007,vol. 48,p. 2353 - 2356
[4]Patent: WO2015/127548,2015,A1 .Location in patent: Paragraph 00156
[5]Patent: WO2016/198585,2016,A1 .Location in patent: Page/Page column 26
- 7
-
5-chloro-2-iodo-4-methoxycarbonyl-benzenediazonium; chloride
[ No CAS ]
-
[ 256935-85-0 ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: NaOEt / ethanol / 2 h / 0 - 5 °C
2: HCl (g) / ethanol |
|
Reference:
[1]Chen, Yihui; Shibata, Masayuki; Rajeswaran, Manju; Srikrishnan, Thamarapu; Dugar, Sundeep; Pandey, Ravindra K.
[Tetrahedron Letters, 2007, vol. 48, # 13, p. 2353 - 2356]
- 8
-
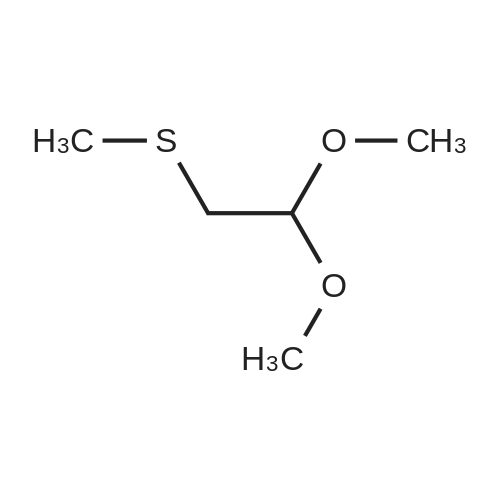
 [ 2457-76-3 ]
[ 2457-76-3 ]
-
[ 256935-85-0 ]
Reference:
[1]Tetrahedron Letters,2007,vol. 48,p. 2353 - 2356
[2]Tetrahedron Letters,2007,vol. 48,p. 2353 - 2356
[3]Patent: WO2014/97140,2014,A1
[4]Patent: WO2015/127548,2015,A1
- 9
-
[ 256935-85-0 ]
-
[ 75-36-5 ]
-
4-acetylamino-5-iodo-2-chloro-benzoic acid methyl ester
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 81% |
With pyridine In tetrahydrofuran at 0℃; for 4h; |
3.B
To a solution of 4-amino-5-iodo-2-chloro-benzoic acid methyl ester (104 g, 330 mmol) in 330 mL THF was added pyridine (40 mL). The solution was cooled to 0° C. and acetyl chloride (35 mL) was added slowly. Stirring was continued for 4 h. The mix was cooled and H2O (800 mL) was added dropwise. Filtered and washed with H2O followed by MeOH/H2O (1:1). Upon drying, the product was obtained in 81% yield. M+H+(354). |
| 78% |
With pyridine In tetrahydrofuran at 10 - 30℃; for 4h; Industry scale; |
1; 4
Example 1 Synthesis of 2-Chloro-4-acetamido-5-iodobenzoic Acid Methyl Ester[0058] The methyl ester of 2-chloro-4-amino-5-iodobenzoic acid (ca. 362 kg) was mixed with tetrahydrofuran (THF: 826 kg) and stirred. Pyridine (138 kg) and more THF (92 kg) were added, and the mixture was cooled to 10-200C. Acetyl chloride (119 kg) was added at a rate that permitted the temperature to be maintained below 250C; THF (92 kg) was used to rinse the acetyl chloride into the reaction vessel. The reaction mixture was stirred for 4 hours or until at least 97% consumption of starting material was achieved as judged by HPLC analysis.[0059] The mixture was cooled to 10-150C, and water (2520 L) was added in 3 batches, while the temperature was maintained at 10-250C. The solids were then collected by processing the mixture in five batches with a centrifuge: each batch was washed with water (91 L), water (91 L), and methanol (91 L). After this, each batch was suspended in methanol (2500 L) and refluxed for one hour, and was then cooled to 0-5 0C. Product was collected by centrifugation and dried at 35-45 0C. If analysis of a batch shows a significant amount of the regioisomeric iodide or the diiodide carried through from the iodination step (e.g., if the product constitutes less than about 93% of the material detected by HPLC), the solids may be slurried with cold methanol and re-centrifuged to remove much of those impurities.[0060] Repeating this reaction at about this scale several times provided a reproducible yield of 78-81%, and the product wasan off-white to light brown or light pink solid that was 97-99% homogenous by HPLC.; Example 4Stage 3 Stage 3 aC8H7ClINO2 C10H9CIINO3 F. W. 311.50 F.W. 353.54 SCI-0216433 SCI-0216434[0071] The Stage 3a process has undergone a number of changes since Campaign S2. These are summarised briefly below:[0072] For Campaigns S2 and S3, an optional methanol re-slurry was included in the Stage 3 a process, dependent on the HPLC purity of the crude wet-cake. If the wet-cake purity was below 93 area % then the re-slurry was performed. This was the case for both S2 and S3 and it was decided that the re-slurry should be made mandatory for S4 for the sake of process consistency. In the event, the crude Stage 3 a product was much purer than in previous campaigns (97.6 area %) and the re-slurry upgraded it to 99.9 area %. Mass balance studies indicated that the re-slurry resulted in a yield loss of around 5 %.[0073] For Campaign S5, the Stage 3a process was transferred to the RPS Dudley One Semi-works facility. The batch size (Stage 3 input) was increased by a factor of 2.3 from 127 to 292 kg. Isolation of the product was carried out using a peeler centrifuge in place of the ceramic vat-filter employed in the Pilot Campaigns.[0074] As the S5 product was to be isolated on a centrifuge, it was anticipated that the crude material would have a greater purity than that seen in earlier campaigns and that the methanol re-slurry would therefore not be required. This proved to be the case and all the crude wet-cake from both batches was found to have purity in excess of 93 area %. In P A T E N T Attorney Docket: SCI5249WOPCT fact, eight out often of the centrifuge baskets had purities greater than 98 %. Centrifugation was clearly more efficient at liquor removal than vat-filtration.[0075] The batch size (Stage 3a input) for Campaign S6 was increased by a factor of 1.31 from 292 to 382 kg. The only other change was the incorporation of additional cold methanol centrifuge washes for one of the batches, which had an abnormally high regioisomer content in the Stage 3 input. This strategy proved successful as the regioisomer content was reduced from as high as 12.9 area % in the Stage 3 input to less than 1 area % in the Stage 3 a product.Comparison of Yield and Purity for Campaigns S3 to S6.[0076] Table One gives a comparison of yield and purity data for Campaigns S3 to S6.Table One: Stage 3a Yield and Purity Data for Campaigns S3 to S6[0077] The assay-based yields have remained fairly constant across the four campaigns S3 to S6. Laboratory experiments indicate that yields in excess of 90 % should be achieved in the P A T E N T Attorney Docket: SCI5249WOPCT αu&cuι.c ui me meuianoi re-smrry. campaigns S3 and S4 both incorporated the re-slurry, which was undoubtedly the main reason for the low yields observed.[0078] Surprisingly, no improvement in yield was seen in S5, which did not incorporate the re- slurry. Due to the relatively high minimum stir volume in the reaction vessel V3166, it proved necessary to charge additional THF (29 % of original charge) in order to achieve efficient agitation. Although the water quench charge was increased by the same proportion, this change resulted in the Stage 3 a product being isolated from a significantly increased solvent volume. This is probably the main reason for the yield short-fall in S5. However, analysis of the mother liquors did not indicate the presence of significant amounts of product. It has since been shown that the assay method underestimates the amount of Stage 3a present when applied to solutions. In future, liquor samples should be concentrated to dryness before attempting to perform an assay.[0079] The S6 yield was even more disappointing than that obtained in S5. There are two principle reasons for this:[0080] The Stage 3 input for the second batch had an abnormally high level of the regioisomeric impurity (up to 12.9 area %). In order to remove this, additional cold methanol washes were applied during centrifugation of the Stage 3 a product. These proved highly effective in removing the impurity, but undoubtedly had a detrimental effect on yield.[0081] For batch 80012941, additional THF had to be charged in order to mobilise solids remaining in vessel V3166 after transfer of the main batch to VlOOl. The solids were successfully mobilised, but it was necessary to charge additional water as an anti-solvent to compensate for the extra THF. In spite of the additional water charge, it is likely that this deviation will have resulted in some loss of product to the liquors.[0082] To prevent a repetition of the losses observed in S5 and S6, the following measures are proposed or have already been implemented:[0083] Particular attention should be paid to isolation of the Stage 3 product on the centrifuge. Efficient de-liquoring at this point is essential to removing the unwanted regioisomer and thereby avoiding the need for additional methanol washes at Stage 3 a.[0084] Larger agitator blades have been fitted to V3166. The resulting improved agitation should avoid the need to charge additional THF to the reaction mixture as in S5. [0085] The reaction mixture should be quenched with water in V3166 before transfer to VlOOl . This should avoid the problems associated with solids settling out in V3166 during the transfer. Once quenched, the mixture is much more mobile and should be transferred more readily, P A T E N T Attorney Docket: SCI5249WOPCT luυsoj In addition to the above changes, an increased reaction concentration has now been demonstrated at Pilot scale. This will result in a significant improvement in volumetric efficiency and allow a larger batch size to be accommodated. Process Outline[0087] Below is the process outline of the Stage 3a process employed in Campaign S6. Process outlines for Campaigns S3, S4 and S5 are contained in the relevant campaign reports. P A T E N T Attorney Docket: SCI5249WOPCT P A T E N T Attorney Docket: SCI5249WOPCT P A T E N T Attorney Docket: SCI5249WOPCT P A T E N T Attorney Docket: SCI5249WOPCT[0088] Expected dry weight = 333 Kg (81 %)Equipment Train[0089] For Campaigns S3 and S4, Stage 3a manufacture, aqueous work-up and crystallisation were performed in PV351, a 300-gallon glass-lined vessel. The product was isolated in PD351 and PD404, ceramic vat-filters associated with PV351 and dried in PK602, a double cabinet air tray-dryer.[0090] Campaigns S5 and S6 were carried out in the RPS Dudley One Semi-works facility. The reaction chemistry took place in V3166, a 1000-gallon hastelloy reactor. The aqueous quench/crystallisation was performed in VlOOl, a 1000-gallon glass-lined vessel, and the product was isolated on D3353, a hastelloy peeler centrifuge. Drying took place in K3045, a stainless steel vacuum tray dryer.Quality-Critical Parameters[0091] No laboratory work to identify quality-critical parameters has been performed for Stage 3a. The product manufactured in Campaigns S3 to S6 has consistently satisfied specification and no quality-critical parameters have been identified from the processing records. P A T E N T Attorney Docket: SCI5249WOPCT[0092] The quality of the Stage 3a manufactured is largely determined by the quality of the Stage 3 input. In Campaign S3 and one batch of S6, relatively high levels of diiodo-Stage 3 were present in the Stage 3 input. This impurity was partially removed during the Stage 3 a process, but not completely. This is reflected in the low purities for the relevant batches shown in Table One. Clearly, good control over formation of the diiodo impurity at Stage 3 is essential to generating high purity Stage 3a.[0093] The other main impurity observed in Stage 3 a is the Stage 3 a regioisomer. This is the product of acetylation of the corresponding Stage 3 impurity. Efficient centrifugation of the Stage 3 product is the main requirement for controlling the level of this impurity. [0094] As indicated earlier, the Stage 3 a process includes provision for a methanol re-slurry if the wet-cake purity is below 93 area %. In reality, centrifugation results in high purity product and the re-slurry was employed in neither S5 nor S6.Parameters Affecting Yield[0095] In Campaigns S5 and S6, it proved necessary to charge additional THF to the process in order to achieve satisfactory slurry mobility. Although additional water (anti-solvent) was charged pro-rata to compensate for this, it seems likely that the additional solvent resulted in some yield losses. As described above, improved agitation and direct quenching of the batch in V3166 should remove the need for extra THF in future campaigns.[0096] The Stage 3 input for S6 batch 80012941A,B,C contained a high level of the Stage 3 regioisomer. This was acetylated to give the Stage 3a regioisomer and extra methanol centrifuge washes were applied to remove it from the product. These undoubtedly resulted in some yield loss. By ensuring efficient regioisomer removal at Stage 3, it should be possible to prevent such losses in future campaigns.[0097] Conversion of Stage 3 to Stage 3 a is monitored by HPLC analysis of the reaction mixture. Table Two gives a comparison of final in-process levels of Stage 3 and the Stage 3 content of the isolated Stage 3 a product. The current in-process limit for unreacted Stage 3 is 3 area % and this has remained unchanged since the process was introduced to the Dudley site in 2001. P A T E N T Attorney Docket: SCI5249WOPCTTable Two: Comparison of In-process and Intermediate Stage 3 levels for Stage 3a Campaigns S3 to S6[0098] The data in Table Two indicate that in-process levels of unreacted Stage 3 up to 2.5 area % are well tolerated by the process. The S6 batch 80012940 gave a final completion result of 2.5 area % Stage 3, but the batch still had less than 1.0 area % of Stage 3 in the isolated product. It would therefore seem that the unreacted starting material is efficiently removed during isolation. From an economic viewpoint, this represents significant lost yield. It is therefore recommended that the in-process limit be lowered to 1 area %. There should be no difficulty in achieving this level. In fact, the lower limit has already been employed successfully in the recent Pilot Trial.Table Three: Comparison of Stage 3a In-process and Intermediate HPLC Purities for CampaignsS3 to S6 P A T E N T Attorney Docket: SCI5249WOPCTa. Re-s urry performed, even though batch met in-process limit, for consistency with previous batches b. Values shown are simple averages for all centrifuge basket-loads c. Values shown are simple averages for all part-batches (A,B etc.) corresponding to oven charges[0099] All batches from Campaign S4 onwards have readily achieved the in-process limit and have therefore not required re-slurry. However, a re-slurry was employed for the S4 batch for the sake of consistency with earlier batches. The Stage 3a product isolated on the centrifuge in S5 and S6 had generally high purity (>98 area %). Batch 80012940 had atypically low purity (96.6 area %) on account of a high level of the diiodo impurity carried over from Stage 3. The high purity of batch 80012941 was ensured by employing additional methanol centrifuge washes to remove the regioisomeric impurity, which was present at high levels due to inefficient centrifugation of the Stage 3 input.[00100] With efficient centrifugation and good control over formation of the diiodo impurity at Stage 3, it should be possible to generate Stage 3a product with greater than 98 area % purity. Up to and including Campaign S6, the Stage 3a purity specification was set very low at 'NLT 85 area %'. It has since been tightened to 'NLT 94 area %'. The methanol re-slurry is known to be effective in upgrading the Stage 3a purity by removing the regioisomeric impurity and, to a lesser extent, the diiodo impurity. It would therefore be prudent to retain the in-process analysis, raising the limit to 'NLT 94 area %'. This would allow material which failed to meet the purity specification to be upgraded before drying.Table Four: Comparison of Stage 3a In-process and Intermediate LOD Values for Campaigns S3 to S6 P A T E N T Attorney Docket: SCI5249WOPCTa. In-process sample was composite of parts A and B[00101] Table Four shows good agreement between in-process and intermediate LOD values for Stage 3a. Based on this data, the current in-process limit is judged adequate to ensure that the intermediate specification (NMT 0.5 % w/w) is achieved.Stage 3a HPLC Profile[00102] Table Five shows the Stage 3a HPLC profiles for Campaigns S3 to S6. The overall purity has generally been greater than 94 area %. In fact, when good control over the regioisomer and diiodo impurities is achieved, purity values in excess of 98 area % have been consistently observed. The overall purity of the Stage 3 a product is less important than the levels of the individual impurities. For example, it is known that the diiodo impurity up to a level of 5.8 area % is removed at Stage 4. hi contrast, there is no data to support onward processing of such high levels of the Stage 3 a regioisomer.Stage 3 a Regioisomer[00103] This impurity is formed by acetylation of the 3-iodo regioisomer foϖned as a major side- product at Stage 3. The level of the impurity is therefore determined by the amount of the 3-iodo compound present in the Stage 3 input. This, in turn, is determined by the efficiency of liquor removal and washing on the centrifuge. Good centrifuge technique at Stage 3 is therefore essential to controlling this impurity. If the impurity is detected at high levels in the Stage 3 product, it can be removed by means of an isopropyl alcohol re-slurry. It can also be removed at Stage 3a, by re-slurrying the wet-cake in methanol. P A T E N T Attorney Docket: SCI5249WOPCTDiiodo Impurity[00104] This impurity is generated at Stage 3 as a result of over-iodination. As yet, there is no proven method for removing it, although the Stage 3a methanol re-slurry is partially effective. Levels up to 5.8 area % have been tolerated by the Stage 4 process (Campaign S3). It is thought that the impurity is converted to the bis(TMS acetylene) compound under the Stage 4 conditions and that this is removed during the crystallisation. The current specification is 'Report value'.RRTs 0.83 and 1.09[00105] These impurities have only been detected in two part-batches. Their identity is not known.'Table Five: Stage 3a HPLC Profiles for Campaigns S3 to S6 |
| 73.6% |
With pyridine In tetrahydrofuran at 10 - 30℃; for 4h; |
1; 4.4
EXAMPLE 1; Synthesis of 2-Chloro-4-acetamido-5-iodobenzoic Acid Methyl Ester; The methyl ester of 2-chloro-4-amino-5-iodobenzoic acid (ca. 362 kg) was mixed with tetrahydrofuran (THF: 826 kg) and stirred. Pyridine (138 kg) and more THF (92 kg) were added, and the mixture was cooled to 10-20° C. Acetyl chloride (119 kg) was added at a rate that permitted the temperature to be maintained below 25° C.; THF (92 kg) was used to rinse the acetyl chloride into the reaction vessel. The reaction mixture was stirred for 4 hours or until at least 97% consumption of starting material was achieved as judged by HPLC analysis. The mixture was cooled to 10-15° C., and water (2520 L) was added in 3 batches, while the temperature was maintained at 10-25° C. The solids were then collected by processing the mixture in five batches with a centrifuge: each batch was washed with water (91 L), water (91 L), and methanol (91 L). After this, each batch was suspended in methanol (2500 L) and refluxed for one hour, and was then cooled to 0-5° C. Product was collected by centrifugation and dried at 35-45° C. If analysis of a batch shows a significant amount of the regioisomeric iodide or the diiodide carried through from the iodination step (e.g., if the product constitutes less than about 93% of the material detected by HPLC), the solids may be slurried with cold methanol and re-centrifuged to remove much of those impurities. Repeating this reaction at about this scale several times provided a reproducible yield of 78-81%, and the product was an off-white to light brown or light pink solid that was 97-99% homogenous by HPLC.; EXAMPLE 4; The Stage 3a process has undergone a number of changes since Campaign S2. These are summarised briefly below: For Campaigns S2 and S3, an optional methanol re-slurry was included in the Stage 3a process, dependent on the HPLC purity of the crude wet-cake. If the wet-cake purity was below 93 area % then the re-slurry was performed. This was the case for both S2 and S3 and it was decided that the re-slurry should be made mandatory for S4 for the sake of process consistency. In the event, the crude Stage 3a product was much purer than in previous campaigns (97.6 area %) and the re-slurry upgraded it to 99.9 area %. Mass balance studies indicated that the re-slurry resulted in a yield loss of around 5%. For Campaign S5, the Stage 3a process was transferred to the RPS Dudley One Semi-works facility. The batch size (Stage 3 input) was increased by a factor of 2.3 from 127 to 292 kg. Isolation of the product was carried out using a peeler centrifuge in place of the ceramic vat-filter employed in the Pilot Campaigns. As the S5 product was to be isolated on a centrifuge, it was anticipated that the crude material would have a greater purity than that seen in earlier campaigns and that the methanol re-slurry would therefore not be required. This proved to be the case and all the crude wet-cake from both batches was found to have purity in excess of 93 area %. In fact, eight out of ten of the centrifuge baskets had purities greater than 98%. Centrifugation was clearly more efficient at liquor removal than vat-filtration. The batch size (Stage 3a input) for Campaign S6 was increased by a factor of 1.31 from 292 to 382 kg. The only other change was the incorporation of additional cold methanol centrifuge washes for one of the batches, which had an abnormally high regioisomer content in the Stage 3 input. This strategy proved successful as the regioisomer content was reduced from as high as 12.9 area % in the Stage 3 input to less than 1 area % in the Stage 3a product. Comparison of Yield and Purity for Campaigns S3 to S6. Table One gives a comparison of yield and purity data for Campaigns S3 to S6. TABLE ONE Stage 3a Yield and Purity Data for Campaigns S3 to S6 Camp'n Stage 3a Camp'n Assay- HPLC Stage 3a Physical based Purity Stage 3a Batch Stage 3 Input Output Yield Yield (Area Campaign Number (kg) (kg) (%) (%) %) S3 80009407A, B 122.7 102.5 73.6 Data not 94.4 available S4 80010873A, B 127.0 114.7 79.6 83.8 99.9 S5 80012347A, B 291.9 274.1 80.8 82.4 99.1 80012348A, B, C 291.9 261.2 98.7 S6 80012940A, B, C 374.0 343.6 78.0 81.6 96.6 80012941A, B, C 391.0 333.8 99.0 The assay-based yields have remained fairly constant across the four campaigns S3 to S6. Laboratory experiments indicate that yields in excess of 90% should be achieved in the absence of the methanol re-slurry. Campaigns S3 and S4 both incorporated the re-slurry, which was undoubtedly the main reason for the low yields observed. Surprisingly, no improvement in yield was seen in S5, which did not incorporate the re-slurry. Due to the relatively high minimum stir volume in the reaction vessel V3166, it proved necessary to charge additional THF (29% of original charge) in order to achieve efficient agitation. Although the water quench charge was increased by the same proportion, this change resulted in the Stage 3a product being isolated from a significantly increased solvent volume. This is probably the main reason for the yield short-fall in S5. However, analysis of the mother liquors did not indicate the presence of significant amounts of product. It has since been shown that the assay method underestimates the amount of Stage 3a present when applied to solutions. In future, liquor samples should be concentrated to dryness before attempting to perform an assay. The S6 yield was even more disappointing than that obtained in S5. There are two principle reasons for this: The Stage 3 input for the second batch had an abnormally high level of the regioisomeric impurity (up to 12.9 area %). In order to remove this, additional cold methanol washes were applied during centrifugation of the Stage 3a product. These proved highly effective in removing the impurity, but undoubtedly had a detrimental effect on yield. For batch 80012941, additional THF had to be charged in order to mobilise solids remaining in vessel V3166 after transfer of the main batch to V1001. The solids were successfully mobilised, but it was necessary to charge additional water as an anti-solvent to compensate for the extra THF. In spite of the additional water charge, it is likely that this deviation will have resulted in some loss of product to the liquors. To prevent a repetition of the losses observed in S5 and S6, the following measures are proposed or have already been implemented: Particular attention should be paid to isolation of the Stage 3 product on the centrifuge. Efficient de-liquoring at this point is essential to removing the unwanted regioisomer and thereby avoiding the need for additional methanol washes at Stage 3a. Larger agitator blades have been fitted to V3166. The resulting improved agitation should avoid the need to charge additional THF to the reaction mixture as in S5. The reaction mixture should be quenched with water in V3166 before transfer to V1001. This should avoid the problems associated with solids settling out in V3166 during the transfer. Once quenched, the mixture is much more mobile and should be transferred more readily, In addition to the above changes, an increased reaction concentration has now been demonstrated at Pilot scale. This will result in a significant improvement in volumetric efficiency and allow a larger batch size to be accommodated. Process Outline Below is the process outline of the Stage 3a process employed in Campaign S6. Process outlines for Campaigns S3, S4 and S5 are contained in the relevant campaign reports. Step Description 1. Charge N-methyl indole acid Stage 3 (362 Kg) to vessel V3166 via the solids charging chute. 2. Charge tetrahydrofuran (56 Kg) to vessel V3166 via solids charge chute as a rinse. 3. Charge tetrahydrofuran (770 Kg) to vessel V3166 via drums. 4. Start V3166 agitator. 5. Charge pyridine (138 Kg) to vessel V3166 via drums. 6. Charge tetrahydrofuran (92 Kg) to vessel V3166, via drums, as a line-wash. 7. Cool contents of vessel V3166 to 10-20° C. 8. Charge acetyl chloride (119 Kg) to dispenser R3173. 9. Transfer contents of dispenser R3173 to vessel V3166 at a rate that maintains the batch temperature in the range 10 to 25° C. 10. Charge tetrahydrofuran (92 Kg) to vessel V3166, via dispenser R3173, as a line wash. 11. Stir contents of vessel V3166 at 20 to 30° C. for NLT 4 hours. 12. Sample contents of vessel V3166 for completion (DI/SD469/306). Limit: NMT 3% Stage 3 by HPLC area %. If complete, continue processing at Step 21. If incomplete, continue processing at Step 13. 13. Stir contents of vessel V3166 at 20 to 30° C. for NLT 2 hours. 14. Sample contents of vessel V3166 for completion (DI/SD469/306). Limit: NMT 3% Stage 3 by HPLC area %. If complete, continue processing at Step 21. If incomplete, continue processing at Step 15. 15. Charge pyridine (quantity specified by chemist) to vessel V3166 via dispenser R3173 maintaining the batch temperature in the range 20 to 30° C. 16. Charge tetrahydrofuran (45 Kg) to vessel V3166 via dispenser R3173. 17. Charge acetyl chloride (quantity specified by chemist) to vessel V3166 via dispenser R3173 maintaining the batch temperature in the range 20 to 30° C. 18. Charge tetrahydrofuran (45 Kg) to vessel V3166 via dispenser R3173. 19. Stir contents of vessel V3166 at 20 to 30° C. for NLT 2 hours. 20. Sample contents of vessel V3166 for completion (DI/SD469/306). Limit: NMT 3% Stage 3 by HPLC area %. If complete, continue processing at Step 21. If incomplete, consult chemist. 21. Transfer contents of vessel V3166 to vessel V1001. 22. Charge tetrahydrofuran (92 Kg) to vessel V3166 via drums. 23. Transfer contents of vessel V3166 to vessel V1001 as a line rinse. 24. Cool contents of vessel V1001 to 10 to 15° C. 25. During the cool back, charge water (631 L) to vessel V1 via bulk supply. 26. Charge contents of V1 to vessel V1001 over NET 1 hour, maintaining the batch temperature in the range 10 to 25° C. 27. Charge water (1261 L) to vessel V3166 via bulk supply and stir for NLT 30 minutes. 28. Transfer contents of V3166 to vessel V1001 maintaining the batch temperature in the range 10 to 25° C. 29. Charge water (631 L) to vessel V3166 via bulk supply and stir for NLT 30 minutes. 30. Transfer contents of V3166 to vessel V1001 maintaining the batch temperature in the range 10 to 25° C. 31. Agitate contents of vessel V1001 at 20-25° C. for NLT 2 hours. 32. Transfer approximately one fifth of the contents of vessel V1001 to centrifuge D3353. Collect liquors in receiver R3350. 33. Charge water (91 L) to header A3160 via drums. 34. Transfer contents of header A3160 to centrifuge D3353 as a cake wash, collecting liquors in receiver R3350. 35. Charge water (91 L) to header A3160 via drums. 36. Charge methanol (91 L) to header A3160 via bulk supply. Circulate to mix. 37. Transfer contents of header A3160 to centrifuge D3353 as a cake wash, collecting liquors in receiver R3350. 38. Drum up contents of receiver R3350 for disposal. 39. Discharge solids from centrifuge D3353 to kegs. 40. Repeat steps 32 to 39 three more times. 41. Transfer remaining contents of vessel V1001 to centrifuge D3353. Collect liquors in receiver R3350. 42. Charge water (91 L) to vessel V1001 via drums. 43. Transfer contents of vessel V1001 to centrifuge D3353 as a cake wash and line rinse, collecting liquors in receiver R3350. 44. Charge water (91 L) to header A3160 via drums. 45. Charge methanol (91 L) to header A3160 via bulk supply. Circulate to mix. 46. Transfer contents of header A3160 to centrifuge D3353 as a cake wash, collecting liquors in receiver R3350. 47. Drum up contents of receiver R3350 for disposal. 48. Discharge solids from centrifuge D3353 to kegs. 49. Sample wet cake for purity. (DI/SD469/307). Limit: NLT 93 area % Stage 3a If the analysis passes, continue at Step 83. If the analysis fails, and this is the first batch of the campaign, continue at step 50. If the analysis fails, and this is the second batch of the campaign, continue at step 50, but omit steps 74-82. 50. Charge wet cake to vessel V3166 via the charge chute. 51. Charge methanol (204 L, 1 full drum) to vessel V3166 via solids charge chute as a line rinse. 52. Charge bulk methanol (1453 L) to vessel V3166. 53. Start agitator and stir contents of V3166 for at least 15 minutes. 54. Transfer contents of V3166 to V1001. 55. Start V1001 agitator. 56. Charge bulk methanol (854 L) to vessel V3166, and stir for at least 15 minutes. 57. Transfer contents of V3166 to V1001 as vessel rinse. 58. Heat contents of vessel V1001 to reflux (65-67° C.). 59. Stir contents of vessel V1001 at reflux for NLT 1 hour. 60. Cool contents of vessel V1001 to 0 to 5° C. over about 2 hours. 61. Stir contents of vessel V1001 at 0 to 5° C. for NLT 1 hour. 62. During coolback charge bulk methanol (700 L) to header A3160 63. Cool contents of header A3160 to 0 to 5° C. 64. Transfer approximately one fifth of the contents of vessel V1001 to centrifuge D3353. Collect liquors in receiver R3350. 65. Transfer methanol (approximately 130 L) from header A3160 to centrifuge D3353 as a cake wash, collecting the liquors in receiver R3350. 66. Spin dry and discharge solids from centrifuge D3353 to kegs. 67. Repeat steps 64 to 66 three more times 68. Transfer remaining contents of vessel V1001 to centrifuge D3353. Collect liquors in receiver R3350. 69. Charge methanol (130 L) to vessel V1001 via bulk supply. 70. Cool contents of vessel V1001 to 0 to 5° C. 71. Transfer the contents of vessel V1001 to centrifuge D3353 as a cake wash and line rinse, collecting the liquors in receiver R3350. 72. Spin dry and discharge solids from centrifuge D3353 to kegs. 73. Transfer remaining contents of header A3160 directly to receiver R3350 and drum up R3350 contents for disposal. 74. V3166 Boil-out Between Batches Charge tetrahydrofuran (573 L -3 full drums) to vessel V3166 via drums. 75. Start agitator and set contents of vessel V3166 to reflux. 76. Allow contents of vessel V3166 to reflux for NLT 1 hour. 77. Cool contents of vessel V3166 to 25 to 30° C. 78. Transfer contents of vessel V3166 to vessel V1001. 79. Bake vessel V3166 dry under vacuum 80. Transfer contents of V1001 to receiver R3350. 81. Cool contents of vessel V1001 to 15 to 25° C. 82. Drum up contents of receiver R3350 for disposal. 83. Charge wet-cake to dryer K3045, keeping baskets separate. 84. Dry wet-cake in dryer K3045 at 35 to 45° C. until an LOD of NMT 0.5% is achieved. 85. Discharge product from dryer K3045 to drums. Expected dry weight = 333 Kg (81%) Equipment Train For Campaigns S3 and S4, Stage 3a manufacture, aqueous work-up and crystallisation were performed in PV351, a 300-gallon glass-lined vessel. The product was isolated in PD351 and PD404, ceramic vat-filters associated with PV351 and dried in PK602, a double cabinet air tray-dryer. Campaigns S5 and S6 were carried out in the RPS Dudley One Semi-works facility. The reaction chemistry took place in V3166, a 1000-gallon hastelloy reactor. The aqueous quench/crystallisation was performed in V1001, a 1000-gallon glass-lined vessel, and the product was isolated on D3353, a hastelloy peeler centrifuge. Drying took place in K3045, a stainless steel vacuum tray dryer. Quality-Critical Parameters No laboratory work to identify quality-critical parameters has been performed for Stage 3a. The product manufactured in Campaigns S3 to S6 has consistently satisfied specification and no quality-critical parameters have been identified from the processing records. The quality of the Stage 3a manufactured is largely determined by the quality of the Stage 3 input. In Campaign S3 and one batch of S6, relatively high levels of diiodo-Stage 3 were present in the Stage 3 input. This impurity was partially removed during the Stage 3a process, but not completely. This is reflected in the low purities for the relevant batches shown in Table One. Clearly, good control over formation of the diiodo impurity at Stage 3 is essential to generating high purity Stage 3a. The other main impurity observed in Stage 3a is the Stage 3a regioisomer. This is the product of acetylation of the corresponding Stage 3 impurity. Efficient centrifugation of the Stage 3 product is the main requirement for controlling the level of this impurity. As indicated earlier, the Stage 3a process includes provision for a methanol re-slurry if the wet-cake purity is below 93 area %. In reality, centrifugation results in high purity product and the re-slurry was employed in neither S5 nor S6. Parameters Affecting Yield In Campaigns S5 and S6, it proved necessary to charge additional THF to the process in order to achieve satisfactory slurry mobility. Although additional water (anti-solvent) was charged pro-rata to compensate for this, it seems likely that the additional solvent resulted in some yield losses. As described above, improved agitation and direct quenching of the batch in V3166 should remove the need for extra THF in future campaigns. The Stage 3 input for S6 batch 80012941A,B,C contained a high level of the Stage 3 regioisomer. This was acetylated to give the Stage 3a regioisomer and extra methanol centrifuge washes were applied to remove it from the product. These undoubtedly resulted in some yield loss. By ensuring efficient regioisomer removal at Stage 3, it should be possible to prevent such losses in future campaigns. Conversion of Stage 3 to Stage 3a is monitored by HPLC analysis of the reaction mixture. Table Two gives a comparison of final in-process levels of Stage 3 and the Stage 3 content of the isolated Stage 3a product. The current in-process limit for unreacted Stage 3 is 3 area % and this has remained unchanged since the process was introduced to the Dudley site in 2001. TABLE TWO Comparison of In-process and Intermediate Stage 3 levels for Stage 3a Campaigns S3 to S6 Final In-process Stage 3 in Isolated Stage 3 Stage 3a Campaign Batch (HPLC Area %) (HPLC Area %) S3 80009407A, B 1.0 A - ND B - ND S4 80010873A, B ND A - ND B - ND S5 80012347A, B ND A - ND B - ND 80012348A, B, C ND A - ND B - ND C - 0.9 S6 80012940A, B, C 2.5 A - ND B - ND C - 0.9 80012941A, B, C 0.3 A - ND B - ND C - 0.2 The data in Table Two indicate that in-process levels of unreacted Stage 3 up to 2.5 area % are well tolerated by the process. The S6 batch 80012940 gave a final completion result of 2.5 area % Stage 3, but the batch still had less than 1.0 area % of Stage 3 in the isolated product. It would therefore seem that the unreacted starting material is efficiently removed during isolation. From an economic viewpoint, this represents significant lost yield. It is therefore recommended that the in-process limit be lowered to 1 area %. There should be no difficulty in achieving this level. In fact, the lower limit has already been employed successfully in the recent Pilot Trial. TABLE THREE Comparison of Stage 3a In-process and Intermediate HPLC Purities for Campaigns S3 to S6 In-Process Intermediate Methanol HPLC Purity HPLC Purity Re-slurry Campaign Batch (Area %) (Area %) Employed? S3 80009407A, B 89.4 94.4 Yes S4 80010873A, B 97.6 99.9Yesa S5 80012347A, B99.0b 99.1c No 80012348A, B, C97.5b 98.7c No S6 80012940A, B, C96.8b 96.6c No 80012941A, B, C99.3b 99.0c No aRe-slurry performed, even though batch met in-process limit, for consistency with previous batches bValues shown are simple averages for all centrifuge basket-loads cValues shown are simple averages for all part-batches (A, B etc.) corresponding to oven charges All batches from Campaign S4 onwards have readily achieved the in-process limit and have therefore not required re-slurry. However, a re-slurry was employed for the S4 batch for the sake of consistency with earlier batches. The Stage 3a product isolated on the centrifuge in S5 and S6 had generally high purity (>98 area %). Batch 80012940 had atypically low purity (96.6 area %) on account of a high level of the diiodo impurity carried over from Stage 3. The high purity of batch 80012941 was ensured by employing additional methanol centrifuge washes to remove the regioisomeric impurity, which was present at high levels due to inefficient centrifugation of the Stage 3 input. With efficient centrifugation and good control over formation of the diiodo impurity at Stage 3, it should be possible to generate Stage 3a product with greater than 98 area % purity. Up to and including Campaign S6, the Stage 3a purity specification was set very low at ‘NLT 85 area %’. It has since been tightened to ‘NLT 94 area %’. The methanol re-slurry is known to be effective in upgrading the Stage 3a purity by removing the regioisomeric impurity and, to a lesser extent, the diiodo impurity. It would therefore be prudent to retain the in-process analysis, raising the limit to ‘NLT 94 area %’. This would allow material which failed to meet the purity specification to be upgraded before drying. TABLE FOUR Comparison of Stage 3a In-process and Intermediate LOD Values for Campaigns S3 to S6 Final In-process Intermediate LOD Campaign Batch LOD (% w/w) (% w/w) S3 80009407A, B0.2a A - ND B - ND S4 80010873A, B0.1a A - 0.1 B - 0.1 S5 80012347A 0.05 ND 80012347B 0.13 0.02 80012348A 0.14 ND 80012348B 0.12 0.05 80012348C 0.06 0.36 S6 80012940A ND 0.05 80012940B ND 0.04 80012940C 0.1 0.04 80012941A ND 0.04 80012941B 0.1 0.05 80012941A 0.1 0.04 aIn-process sample was composite of parts A and B Table Four shows good agreement between in-process and intermediate LOD values for Stage 3a. Based on this data, the current in-process limit is judged adequate to ensure that the intermediate specification (NMT 0.5% w/w) is achieved. Stage 3a HPLC Profile Table Five shows the Stage 3a HPLC profiles for Campaigns S3 to S6. The overall purity has generally been greater than 94 area %. In fact, when good control over the regioisomer and diiodo impurities is achieved, purity values in excess of 98 area % have been consistently observed. The overall purity of the Stage 3a product is less important than the levels of the individual impurities. For example, it is known that the diiodo impurity up to a level of 5.8 area % is removed at Stage 4. In contrast, there is no data to support onward processing of such high levels of the Stage 3a regioisomer. Stage 3a Regioisomer This impurity is formed by acetylation of the 3-iodo regioisomer formed as a major side-product at Stage 3. The level of the impurity is therefore determined by the amount of the 3-iodo compound present in the Stage 3 input. This, in turn, is determined by the efficiency of liquor removal and washing on the centrifuge. Good centrifuge technique at Stage 3 is therefore essential to controlling this impurity. If the impurity is detected at high levels in the Stage 3 product, it can be removed by means of an isopropyl alcohol re-slurry. It can also be removed at Stage 3a, by re-slurrying the wet-cake in methanol. Diiodo Impurity This impurity is generated at Stage 3 as a result of over-iodination. As yet, there is no proven method for removing it, although the Stage 3a methanol re-slurry is partially effective. Levels up to 5.8 area % have been tolerated by the Stage 4 process (Campaign S3). It is thought that the impurity is converted to the bis(TMS acetylene) compound under the Stage 4 conditions and that this is removed during the crystallisation. The current specification is ‘Report value’. RRTs 0.83 and 1.09 These impurities have only been detected in two part-batches. Their identity is not known.’ TABLE FIVE Stage 3a HPLC Profiles for Campaigns S3 to S6 Component (Area %) Stage 3a RRT Regio- Diiodo Campaign Batch Stage 3 0.83 isomer RRT 1.09 Stage 3a Impurity S3 80009407A ND ND ND ND 94.2 5.8 80009407B ND ND ND ND 94.5 5.5 S4 80010873A ND ND ND ND 100.0 ND 80010873B ND 0.21 <0.1 ND 99.7 ND S5 80012347A ND ND ND ND 99.13 0.87 80012347B ND ND ND ND 99.12 0.88 80012348A ND ND ND ND 99.25 0.75 80012348B ND ND ND ND 99.24 0.76 80012348C 0.9 ND 0.12 ND 97.9 1.1 S6 80012940A ND ND <0.1 ND 97.6 2.4 80012940B ND ND <0.1 ND 97.0 2.9 80012940C 0.9 ND <0.1 0.58 95.3 3.2 80012941A ND ND 0.4 ND 99.6 ND 80012941B ND ND 0.5 ND 99.2 0.7 80012941C ND ND 0.8 ND 98.2 1.0 |
Reference:
[1]Current Patent Assignee: JOHNSON & JOHNSON INC - US2004/142940, 2004, A1
Location in patent: Page/Page column 14
[2]Current Patent Assignee: JOHNSON & JOHNSON INC - WO2008/79105, 2008, A1
Location in patent: Page/Page column 21; 24-36
[3]Current Patent Assignee: JOHNSON & JOHNSON INC - US2007/66833, 2007, A1
Location in patent: Page/Page column 9; 10-15
- 10
-
benzyl trimethylammonium dichloroiodate
[ No CAS ]
-
[ 46004-37-9 ]
-
[ 256935-85-0 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With calcium carbonate; In methanol; dichloromethane; ethyl acetate; |
EXAMPLE 58A methyl 4-amino-2-chloro-5-iodo-benzoate A solution of <strong>[46004-37-9]methyl 4-amino-2-chloro-benzoate</strong> (1.14 g, 6.16 mmol) in dichloromethane (30 mL) and methanol (15 mL) was treated sequentially with calcium carbonate (1.85 g, 18.5 mmol) and benzyl trimethylammonium dichloroiodate (3.22 g, 9.24 mmol), stirred for 18 hours, treated with additional benzyl trimethylammonium dichloroiodate (2 g, 5.75 mmol), stirred for 3 days, filtered into a separatory funnel, washed with saturated NaHSO3, dried (MgSO4), filtered, and concentrated. The concentrate was recrystallized from ethyl acetate to provide the majority of the desired product. The filtrate was concentrated, and the concentrate was purified on silica gel with a gradient of from 10% to 20% ethyl acetate/hexanes to provide a total of 1.3 g of the desired product. MS (ESI(-)) m/z 310 (M-H)-; 1H NMR (CDCl3) d 8.25 (d, J=2.0 Hz, 1H), 6.75 (d, J=2.0 Hz, 1H), 4.52 (br s, 2H), 3.87 (s, 3H). Characterization for the minor product: 1H NMR (300 MHz, CDCl3) 7.72 (d, J=8.8 Hz 1H), 6.63 (d, J=8.5 Hz, 1H), 4.71 (br s, 2H), 3.88 (s, 3H). |
Reference:
[1]Patent: US6228868,2001,B1
- 11
-
[ 256935-85-0 ]
-
catechol borane
[ No CAS ]
-
[ 927-80-0 ]
-
 [ 162100-83-6 ]
[ 162100-83-6 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With hydrogenchloride; sodium hydroxide;tetrakis(triphenylphosphine)palladium (0); In tetrahydrofuran; ethyl acetate; |
EXAMPLE 58B methyl 6-chloro-5-indolecarboxylate A 50% solution of ethyl ethynyl ether in hexanes (2.1 mL, 10.8 mmol) at 0 C. was slowly treated with 1M catechol borane in THF (9.7 mL, 9.7 mmol), warmed to room temperature, stirred for 2 hours, heated at 70 C. for 2 hours, cooled to room temperature, treated sequentially with Example 58A (1.77 g, 5.69 mmol) in THF (30 mL), tetrakis(triphenylphosphine)-palladium(0) (329 mg, 0.28 mmol), and powdered sodium hydroxide (683 mg, 17.1 mmol), heated to reflux, stirred for 18 hours, cooled to room temperature, treated with 2M HCl (30 mL), stirred for 18 hours, treated with ethyl acetate, washed sequentially with water, 2M Na2CO3, and brine, dried (MgSO4), filtered, and concentrated. The concentrate was purified by flash column chromatography on silica gel to provide the impure desired product which was used in the next step without further purification. |
Reference:
[1]Patent: US6228868,2001,B1
- 13
-
[ 256935-85-0 ]
-
 [ 1066-54-2 ]
[ 1066-54-2 ]
-
 [ 1036337-13-9 ]
[ 1036337-13-9 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With triethylamine In tetrahydrofuran at 20℃; for 1.5h; Inert atmosphere; |
23.C
C. Methyl 4-amino-2-chloro-5-((trimethylsilyl)ethynyl)benzoate, 23e. To a mixture of compound 23c (0.642 mmol, 200 mg), CuI (0.064 mmol, 12.2 mg) and Pd(PPh3)2Cl2 (0.064 mmol, 45 mg) in THF (2 mL) was added ethynyltrimethylsilane (0.963 mmol, 95 mg) followed by Et3N (7.19 mmol, 1 mL) under N2. The reaction mixture was stirred at room temperature for 1.5 h and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 15% EtOAc/hexanes) to give compound 23e. |
|
With triethylamine In tetrahydrofuran at 20℃; for 1.5h; Inert atmosphere; |
23.C
To a mixture of compound 23c (0.642 mmol, 200 mg), CuI (0.064 mmol, 12.2 mg) and Pd(PPh3)2Cl2 (0.064 mmol, 45 mg) in THF (2 mL) was added ethynyltrimethylsilane (0.963 mmol, 95 mg) followed by Et3N (7.19 mmol, 1 mL) under N2. The reaction mixture was stirred at room temperature for 1.5 h and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 15% EtOAc/hexanes) to give compound 23e. |
|
With triethylamine In tetrahydrofuran at 20℃; for 1.5h; Inert atmosphere; |
22a.J
To a mixture of compound 22k (200 mg, 0.642 mmol), CuI (12.2 mg, 0.064 mmol) and Pd(PPh3)2Cl2 (45 mg, 0.064 mmol) in THF (2 mL) was added ethynyltrimethylsilane (95 mg, 0.963 mmol) followed by Et3N (1 mL, 7.19 mmol) under N2. The reaction mixture was stirred at room temperature for 1.5 h and then partitioned between EtOAc and water. The organic layer was concentrated and purified by flash column chromatography (silica gel, 15% EtOAc/hexanes) to give compound 22m. |
Reference:
[1]Current Patent Assignee: JOHNSON & JOHNSON INC - US2012/58986, 2012, A1
Location in patent: Page/Page column 34
[2]Current Patent Assignee: JOHNSON & JOHNSON INC - US2012/77797, 2012, A1
Location in patent: Page/Page column 33; 34
[3]Current Patent Assignee: JOHNSON & JOHNSON INC - US2012/101081, 2012, A1
Location in patent: Page/Page column 53
- 35
-
[ 256935-85-0 ]
-
(4-amino-2-chloro-5-iodophenyl)methanol
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 15.5 g |
With diisobutylaluminium hydride In tetrahydrofuran; dichloromethane at -40 - 20℃; for 2.5h; Inert atmosphere; |
1.3 Step 3: (4-amino-2-chloro-5-iodophenyl )methanol
To a solution of methyl 4-amino-2-chloro-5- iodo-benzoate (17.0 g, 54.6 mmol) in CH2CI2 (400 mL) and THE (100 mL) at -40 °C under nitrogen was slowly added diisobutylaluminium hydride (136.0 mL, 136.0 mmol, 1.0 M in CH2CI2) over a time period of 30 mm. The cooling bath was removed on completion of the addition. After stirring at rt for 2 h, the mixture was poured into a 0 °C saturated aqueous potassium sodium tartrate. The layers were partitioned and the aqueous layer was extracted with CH2CI2 (600 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated to dryness under vacuum to afford 15.5 g of title compound as an orange solid which was used in next step without further purification. |
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
Location in patent: Paragraph 00157
- 36
-
[ 256935-85-0 ]
-
4-amino-2-chloro-5-iodobenzaldehyde
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 37
-
[ 256935-85-0 ]
-
ethyl 2-((5-amino-2-formyl-4-iodophenyl)thio)acetate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 3 steps
1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3: potassium carbonate / N,N-dimethyl-formamide / 72 h / 20 °C / Inert atmosphere |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 38
-
[ 256935-85-0 ]
-
ethyl 6-amino-5-iodobenzo[b]thiophene-2-carboxylate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 3 steps
1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 39
-
[ 256935-85-0 ]
-
ethyl 6-bromo-5-iodobenzo[b]thiophene-2-carboxylate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 4 steps
1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 40
-
[ 256935-85-0 ]
-
ethyl 6-bromo-5-((diethoxyphosphoryl)difluoromethyl)benzo[b]thiophene-2-carboxylate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 5 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 41
-
[ 256935-85-0 ]
-
bis-ammonium((6-bromo-2-(ethoxycarbonyl)benzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 6 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: trimethylsilyl bromide / dichloromethane / 20 °C
6.2: 0.5 h |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 42
-
[ 256935-85-0 ]
-
6-bromo-5-((ethoxy(hydroxy)phosphoryl)difluoromethyl)benzo[b]thiophene-2-carboxylic acid
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 6 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: lithium hydroxide; ethanol / tetrahydrofuran / 3 h / 0 - 20 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 43
-
[ 256935-85-0 ]
-
tris-ammonium 6-bromo-5-(difluoro(phosphonato)methyl)benzo[b]thiophene-2-carboxylate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 7 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: lithium hydroxide; ethanol / tetrahydrofuran / 3 h / 0 - 20 °C
7.1: trimethylsilyl bromide / dichloromethane / 20 °C
7.2: 0.5 h |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 44
-
[ 256935-85-0 ]
-
ethyl hydrogen((6-bromo-2-carbamoylbenzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 6 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: ammonium hydroxide / tetrahydrofuran / 65 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 45
-
[ 256935-85-0 ]
-
bis-ammonium((6-bromo-2-carbamoylbenzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 7 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: ammonium hydroxide / tetrahydrofuran / 65 °C
7.1: trimethylsilyl bromide / dichloromethane / 20 °C
7.2: 0.5 h |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 46
-
[ 256935-85-0 ]
-
ethyl methyl((6-bromo-2-cyanobenzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 7 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: ammonium hydroxide / tetrahydrofuran / 65 °C
7.1: trichlorophosphate / 90 °C
7.2: 0.5 h / 20 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 47
-
[ 256935-85-0 ]
-
bis-ammonium((6-bromo-2-cyanobenzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 8 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: ammonium hydroxide / tetrahydrofuran / 65 °C
7.1: trichlorophosphate / 90 °C
7.2: 0.5 h / 20 °C
8.1: trimethylsilyl bromide / dichloromethane / 20 °C
8.2: 0.5 h |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 48
-
[ 256935-85-0 ]
-
diethyl((6-bromo-2-(hydroxymethyl)benzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 6 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: sodium tetrahydroborate; methanol / tetrahydrofuran / 3.5 h / 65 °C |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 49
-
[ 256935-85-0 ]
-
bis-ammonium((6-bromo-2-(hydroxymethyl)benzo[b]thiophen-5-yl)difluoromethyl)phosphonate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 7 steps
1.1: diisobutylaluminium hydride / dichloromethane; tetrahydrofuran / 2.5 h / -40 - 20 °C / Inert atmosphere
2.1: manganese(IV) oxide / N,N-dimethyl-formamide / 36 h / 20 °C
3.1: potassium carbonate / N,N-dimethyl-formamide / 77 h / 20 - 75 °C / Inert atmosphere
4.1: copper(I) bromide; tert.-butylnitrite / acetonitrile / 6 h / 45 °C
5.1: copper(I) bromide / tetrahydrofuran / 0.5 h / 20 °C
5.2: 45 °C
6.1: sodium tetrahydroborate; methanol / tetrahydrofuran / 3.5 h / 65 °C
7.1: trimethylsilyl bromide / dichloromethane / 20 °C
7.2: 0.5 h |
|
Reference:
[1]Current Patent Assignee: MCGILL UNIVERSITY - WO2015/127548, 2015, A1
- 50
-
[ 256935-85-0 ]
-
 [ 50978-45-5 ]
[ 50978-45-5 ]
-
methyl 4-amino-2-chloro-5-formylbenzoate
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 45% |
Stage #1: methyl 4-amino-2-chloro-5-iodo-benzoate; 3-oxobenzo[d]isothiazole-2(3H)-carbaldehyde 1,1-dioxide With triethylsilane; 1,1'-bis-(diphenylphosphino)ferrocene; palladium diacetate; 1,4-di(diphenylphosphino)-butane In N,N-dimethyl-formamide at 20℃; for 0.0833333h; Inert atmosphere; Sealed tube;
Stage #2: With triethylamine In N,N-dimethyl-formamide at 115℃; for 1h; Inert atmosphere; Sealed tube; |
Methyl 4-amino-2-chloro-5-formylbenzoate (6a).
Methyl 4-amino-2-chloro-5-iodobenzoate (398 mg, 1.28 mmol, Combi-Blocks Inc.), N-formylsaccharin (809 mg, 3.83 mmol, Combi-Blocks Inc.), palladium(ii) acetate (28.7 mg, 0.128 mmol, Umicore AG & Co.KG.), 1,4-bis(diphenylphosphino)butane (109 mg, 0.256 mmol, Sigma-Aldrich Corporation), and triethylsilane (193 mg, 0.265 mL, 1.661 mmol, Sigma-Aldrich Corporation) were mixed in N, N-dimethylformamide (3 mL) in a sealed vial under a nitrogen atmosphere. The mixture was stirred at RT for 5 min. The stirring was stopped, and triethylamine (388 mg, 0.539 mL, 3.83 mmol) was added carefully to avoid as much mixing as possible (bubbling observed). The reaction mixture was heated at 115°C for 1 h then cooled to RT and partitioned between EtOAc (80 mL) and saturated NH4Cl (60 mL). The organic layer was separated, washed with brine (50 mL), dried over MgSO4, filtered, and concentrated in vacuo. Chromatographic purification of the residue (silica gel, 0 -30% EtOAc in heptane) gave 6a (122 mg, 0.571 mmol, 45% yield) as a yellow solid. MS (ESI, +ve) m/z: 213.9 [M + H]+.1H NMR (400 MHz, CHLOROFORM-d) δ ppm 9.86 (1 H, s) 8.21 (1 H, s) 6.73 (1 H, s) 3.90 (3 H, s). |
| 0.165 g |
With triethylsilane; palladium diacetate; triethylamine; 1,4-di(diphenylphosphino)-butane In N,N-dimethyl-formamide at 115℃; for 1h; Microwave irradiation; Inert atmosphere; |
P2.2 Step 2:
Step 2: A mixture of compound 7 (0.500 g, 1.605 mmol), N- formylsaccharin (1.017 g, 4.815 mmol), palladium (II) acetate (0.036 g, 0.161 mmol) and 1,4- butylenebis(diphenylphosphine) (dppb; 0.140 g, 0.321 mmol) was treated with anhydrous N,N-dimethylformamide (3.210 mL) and triethylsilane (0.34 mL, 2.087 mmol) in a sealed 2 - 5 mL microwave vial under a nitrogen atmosphere at room temperature. The resulting mixture was stirred for 5 minutes, then stirring was stopped and triethylamine (0.671 mL, 4.815 mmol) was carefully layered over the reaction mixture [note: some effervescence]. The resulting mixture was stirred at room temperature for 1 min and then heated to 115°C for 1 hour under microwave irradiation. The microwave vial was then carefully vented by piercing the septum with a needle, and the reaction mixture was poured into saturated aqueous sodium hydrogen carbonate solution. This mixture was extracted with ethyl acetate (3x), and the combined ethyl acetate extracts were washed with water (2x) and brine, then dried by passing through a phase-separating cartridge. The filtrate was evaporated under reduced pressure, and the crude residue was purified by flash chromatography (CombiFlash Rf, eluting with 0-20% ethylacetate in hexanes using a 12g silica GOLD column) to afford compound 8 (0.165 g) as a pale yellow solid. 1H NMR (400 MHz, CDC13) δ 9.86 (s, 1H), 8.22 (s, 1H), 6.73 (s, 1H), 3.90 (s, 3H) |
Reference:
[1]Current Patent Assignee: AMGEN INC - WO2021/163344, 2021, A1
Location in patent: Paragraph 0097
[2]Current Patent Assignee: Syngenta (in: Sinochem Holdings); SINOCHEM HOLDINGS CORPORATION LTD - WO2016/198585, 2016, A1
Location in patent: Page/Page column 26-27
 630-580-1088
630-580-1088
 sales@ambeed.com
sales@ambeed.com
 Hello, Sign in
Hello, Sign in

 0
0

 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science
 Contact Us
Contact Us
 0
0




 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping